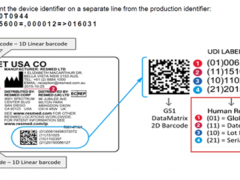
1、什么是突破性器械项目?
突破性器械项目(Breakthrough Devices Program)是一项针对治疗或诊断危害生命或不可逆转的使人衰弱的疾病的医疗急需医疗器械和以器械为主导的组合产品的自愿性项目。
突破性器械项目的目标是根据美国食品药品管理局(以下简称FDA)保护和促进公众健康的使命,在符合上市前批准、510(k)和De Novo的法定标准的同时,通过加快开发、评估和审查,为患者和医疗保健提供者提供及时获得这些器械产品的机会。
突破性器械项目取代了医疗器械加速途径(Expedited Access Pathway)和优先审查(Priority Review),前期获得加速途径指定的医疗器械即被认为符合突破性器械项目。
2、加入突破性器械项目有什么优势?
1)及时有效的沟通交流。制造商可通过多种方式与FDA专家,及时有效地解决上市前审查阶段出现的问题。
2)优先审查。突破性器械项目指南的第II和IV部分具体阐述了突破性器械项目的原则和功能。
3、符合突破性器械项目的条件是什么?
申请上市前批准(PMA)、上市前通告 (510(k)) 或 De Novo指定的器械产品,需同时符合以下两个条件:
1).该器械治疗或诊断危害生命或不可逆转的使人衰弱的疾病。
2).该器械应满足以下至少一项:
a.代表突破性技术,
b.无已批上市替代产品,
c.与现有或已批准的替代产品相比具有显著优势,
d.器械可及性符合患者最大利益。
4、何时申请突破性器械项目指定?
提交上市申请之前的任何时间。
5、如何申请突破性器械项目指定?
通过提交“突破性器械项目指定请求”Q-Submission 来申请突破性器械项目指定。
FDA可发现并建议适合突破性器械项目的候选产品,并向申报者推荐申请该项目。
6、突破性器械项目指定申请中包含的内容?
申请资料应包括产品描述信息、适应症、监管情况、阐述产品满足突破性器械项目的理由,以及您计划的申报途径等。
7、突破性器械项目指定结果的告知方式?
FDA在收到请求后30天内提供提出补充资料要求,以告知突破性设备指定决定,60天内以信函形式告知申报者该产品是否获得突破性器械项目指定。
8、申办者可以从 FDA 获得什么?
获得突破性器械项目指定的产品,可通过多种方式与FDA交流沟通,如讨论设计开发计划和讨论临床试验方案等。并在可能涉及的审评过程中获得优先审评。
9、是否还有其他快速审评途径或项目?
导致不符合突破性器械项目条件的原因,是申报产品不符合该项目定义,即治疗或诊断危害生命或不可逆使人衰弱疾病的医疗急需器械医疗器械和以器械为主导的组合产品。申报者可以考虑申报产品是否符合其他项目,如更安全技术项目(STeP)等。
10、FDA 是否会公示结果?
在获得上市许可之前,除非申报者自愿公开相关信息信息,FDA不会公开披露突破性器械项目指定申请情况和该申请的批准情况。但FDA计划在网页上公开获得上市许可的突破性器械项目产品清单。
11、突破性器械项目相关数据
截至2023年6月30日,医疗器械与放射健康中心(CDRH)和生物制品审评与研究中心(CBER)已授予突破性器械项目指定839个,包括最初在医疗器械加速途径(Expedited Access Pathway)项目产品。获得突破性器械指定的839个产品中,CDRH授予831个,CBER授予8个。
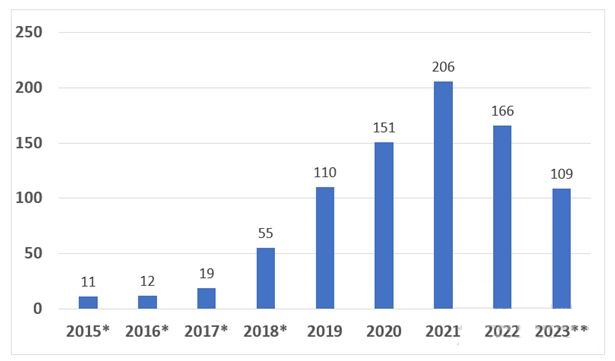
图 1:按年度授予的突破性器械指定数量
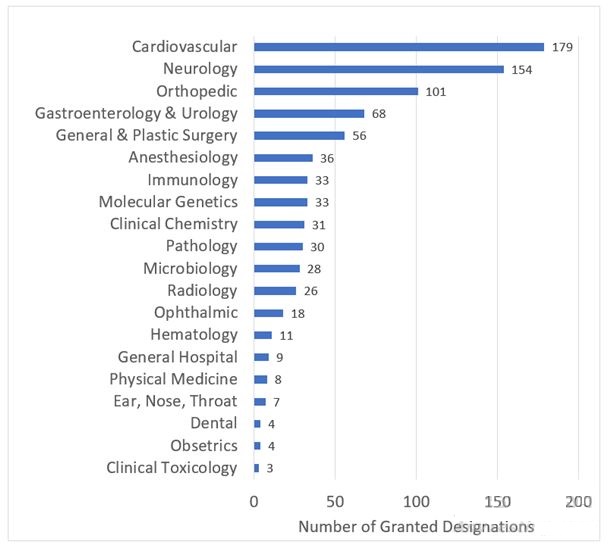
图 2:突破性器械指定按临床使用划分情况
12、CDRH和CBER批准上市的突破性器械数量
共批准上市81个,其中CDRH批准77个,CBER批准4个。