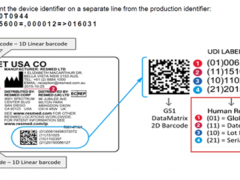
2024年7月9日,IVDR过渡期修正案Regulation (EU)2024/1860正式发布。其中更新后第110条讲述了遗留器械的过渡期时限和可以享受过渡期的条件。针对这份延期修正案,欧盟委员会发布了一份《体外诊断医疗器械法规》(IVDR)过渡期延长相关的常见问题解答(Q&A)。
我们整理了该文件前四个部分的内容,有需要的可以在文末点击“阅读原文”查看详细信息。
PART A – 过渡期延长的范围
1. 哪些器械可以享受延长的过渡期?
IVDR第110条中明确规定了哪些器械可以享受延长的过渡期,在该文件的附录部分提供了一个流程图,我们可以借助该图来决定某个器械是否属于修订后的第110条所涵盖的范围。
2. 已获得IVDR认证的IVDD遗留器械是否还可以享受过渡期?
是的。对于IVDD证书涵盖的遗留器械,只要IVDD证书未被NB撤销,该器械就能享受过渡期。
对于首次需要根据IVDR进行认证的遗留器械,只要其继续满足IVDR第110(3c)条规定的条件,它们就可以在IVDR证书颁发后享受延长过渡期。这就意味着“遗留器械”和相应的符合IVDR的器械可以同时投放市场,直至相关过渡期结束。
PART B – 延长过渡期的证据
1. 制造商如何证明其遗留器械已享受延长的过渡期?
众所周知,制造商可能需要向第三方证明其器械可以合法地投放到欧盟市场或投入使用。为此,制造商应能够使用不同的方式来证明其器械在延长的过渡期内,并且适用是还可以提供有效证书。
制造商应能提供一份自我声明,确认已满足延期条件,并说明过渡期的结束日期。这种自我声明应清楚地标明延期所涉及的器械和所包含的任何证书。Medtech发布的制造商声明模板可供大家参考。
还可以通过NB机构出具的确认函或协议副本来提供额外的证据,表明制造商已提交符合性评估申请并达成书面协议。
主管当局出具的FSC。
PART C – 享受延长过渡期的条件
1. 制造商提出正式申请需具备哪些因素?
在签署书面协议之前,不需要NB对申请进行全面审查。
符合性评估申请不需要包括申请所涵盖的、须接受技术文档审核的每种器械的技术文档。但是,申请必须清楚地表明制造商和申请所涵盖的器械。
制造商在提交申请时应提供可能提交个别技术文件及其他相关信息的时间表。
制造商最迟需要在2025年5月26日之前遵守IVDR的质量管理体系(QMS)要求,QMS符合性评估申请应包括制造商的质量管理体系文件。
2. 如果申请撤回或书面协议终止,会发生什么情况?
若制造商在相关截止日期后撤回申请或终止书面协议,过渡期将不再适用;但若制造商同时与另一个NB签订书面协议并转移申请,且满足其他条件,过渡期将继续适用。
3. 制造商必须提供哪些证据来证明其已根据IVDR建立QMS?
根据IVDR第110(3c)条d点,制造商必须在2025年5月26日之前根据IVDR第10(8)条建立质量管理体系。
根据IVDR第110(3e)条,遵守与上市后监督、市场监督、警戒和注册有关的质量管理体系相关要求是NB适当监督的一部分。
作为IVDR认证的一部分,NB将对整个质量管理体系是否符合IVDR进行评估。
QMS文件需要作为制造商打算转入IVDR的任何器械的符合性评估申请的一部分,并在不迟于以下相关截止日期提交给NB机构:
▶ D类器械和IVDD证书涵盖的器械不得迟于2025年5月26日,
▶ C类器械不得迟于2026年5月26日,
▶ B类和A类无菌器械不得迟于2027年5月26日。