


1、《企业落实医疗器械质量安全主体责任监督管理规定》包含哪些内容?
答:《规定》共六章三十条,主要包括三方面内容:
一是明确质量安全关键岗位要求。生产企业质量安全关键岗位人员包括企业法定代表人和主要负责人、管理者代表、质量管理部门负责人。经营企业质量安全关键岗位负责人员包括企业负责人、质量负责人、质量管理人员,细化各岗位职责和任职条件。
二是规范质量安全管理要求。规定了质量安全管理调度和风险会商制度,细化委托生产管理、产品放行等关键环节管理要求,明确各环节负责人员。
三是制定履职保障机制。要求企业制定质量安全关键岗位说明书并对相关人员进行岗前培训和继续教育,规定关键岗位人员应当在职在岗,明确尽职免责制度和企业对相关人员的奖惩制度。
《规定》施行后,与现行医疗器械生产、经营相关文件不同的条款,按照《规定》要求执行。下一步,国家药监局将继续组织修订医疗器械生产、经营质量管理规范,与《规定》有效衔接。
2、对于专用标准实施日期在2025年12月31日之后的,产品注册备案如何执行新版GB 9706系列标准?
答:对于相关标准发布公告规定的实施日期在2025年12月31日之后的专用标准,在14号通告第二(一)款规定的相关标准实施之日起,首次申请注册或者首次办理备案的产品,应当提交符合新版GB 9706系列标准要求的检验报告。在此之前申请注册并获得受理的,可以按照原标准进行检验、审评审批。例如,对于脉搏血氧仪,其适用专用标准中最后实施的是YY 9706.261-2023《医用电气设备 第2-61部分:脉搏血氧设备的基本安全和基本性能专用要求》(2026年1月15日实施),因此,自2026年1月15日起,首次申请注册的,应当提交符合新版GB 9706系列标准的检验报告。鼓励医疗器械注册人备案人提前实施新版GB 9706系列标准。
3、关于取消创新审查结果告知后申报注册时限延期对于在GB 9706.1-2020及配套并列标准、专用标准(一下简称新版GB 9706系列标准)实施之日前已注册的产品,延续注册如何执行?
答:《关于GB 9706.1-2020及配套并列标准、专用标准实施有关工作的通告》(2023年第14号,以下简称14号通告),未专门针对延续注册作出特殊规定,延续注册应当符合相关法规、规章及规范性文件要求。
根据《医疗器械监督管理条例》第二十二条、《医疗器械注册与备案管理办法》第八十二、八十三条规定,医疗器械注册证有效期为5年。有效期届满需要延续注册的,应当在有效期届满6个月前向原注册部门提出延续注册申请。有下列情形之一的,不予延续注册:(一)未在规定期限内提出延续注册申请;(二)医疗器械强制性标准已经修订,申请延续注册的医疗器械不能达到新要求;(三)附条件批准的医疗器械,未在规定期限内完成医疗器械注册证载明事项。因此,新的医疗器械强制性标准发布实施后,申请延续注册的医疗器械不能达到新标准要求的,不予延续注册。
《国家药品监督管理局关于实施〈医疗器械注册与备案管理办法〉〈体外诊断试剂注册与备案管理办法〉有关事项的通告》(2021年第76号)明确:“对于申请注册的医疗器械,其产品技术要求中引用的强制性标准发生变化的,除国家药监局在发布实施标准文件中另有规定外,在新标准实施之日前受理注册的,可以按照原标准进行审评审批。自新标准实施之日起,企业应当全面实施新标准,产品应当符合新标准要求”。
14号通告第二(一)款对产品注册备案执行新版GB 9706系列标准进行了明确要求:“产品适用GB 9706.1-2020配套专用标准的,GB 9706.1-2020及配套并列标准可与最后实施的专用标准同步实施。产品无适用GB 9706.1-2020配套专用标准的,GB 9706.1-2020及配套并列标准自2023年5月1日实施。” 因此,在14号通告第二(一)款规定的新版GB 9706系列标准实施日期前申请延续注册的,可按原标准要求审评审批;在14号通告第二(一)款规定的新版GB 9706系列标准实施日期后申请延续注册的,应当按新版GB 9706系列标准要求审评审批。例如,对于无适用专用标准的产品,2023年5月1日前申请的延续注册申请,可按原标准要求审评审批;2023年5月1日起申请的,应当按新版GB 9706系列标准要求审评审批。对于产品有适用的专用标准的,如专用标准发布公告规定的实施日期为2024年5月1日,则2024年5月1日前申请的延续注册申请,可按原标准要求审评审批;2024年5月1日起申请的,应当按新版GB 9706系列标准要求审评审批。延续注册申报资料应当符合《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》(2021年第121号)中附件6“医疗器械延续注册申报资料要求及说明”要求。
建议注册人根据产品注册证有效期、检验工作预期完成情况等,适时申请延续注册。
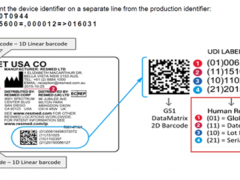
4、什么是医疗器械唯一标识?
答:医疗器械唯一标识由产品标识和生产标识组成,产品标识是识别注册人/备案人、医疗器械型号规格和包装的唯一代码,是从数据库获取医疗器械相关信息的“关键字”,是唯一标识的必须部分;生产标识包括与生产过程相关的信息,包括产品批号、序列号、生产日期和失效日期等,可与产品标识联合使用,满足医疗器械流通和使用环节精细化识别和记录的需求。
唯一标识具备唯一性、稳定性和可扩展性的原则。唯一性是首要原则,是确保产品精确识别的基础,是唯一标识发挥功能的核心原则。由于医疗器械产品的复杂性,唯一性应当与产品识别要求相一致,对于相同特征的医疗器械,唯一性应当指向单个规格型号产品;对于按照批次生产控制的产品,唯一性指向同批次产品;而对于采用序列号生产控制的医疗器械,唯一性应当指向单个产品。
稳定性是指唯一标识一旦分配给医疗器械产品,只要其基本特征没有发生变化,产品标识就应该保持不变。当医疗器械停止销售、使用时,其产品标识不得用于其他医疗器械;重新销售、使用时,可使用原产品标识。
可扩展性是指唯一标识应当与监管要求和实际应用不断发展相适应,“唯一”一词并不意味着对单个产品进行序列号化管理,在唯一标识中,生产标识可以和产品标识联合使用,实现规格型号、批次和单个产品三个层次的唯一性,从而满足当前和未来对医疗器械的识别需求。
5、为什么要建设医疗器械唯一标识系统?
答:医疗技术、药品、医疗器械是构成医疗服务体系的三大支柱,医疗器械涉及声、光、电、磁、图像、材料、力学等行业和近百个专业学科,是国际公认的高新技术产业,具有高技术密集、学科交叉广泛、技术集成融合等特点,代表着一个国家高新技术的综合实力。近年来,医疗器械产业发展迅猛,新技术、新产品层出不穷,产品多样性、复杂性程度不断提升,医疗器械在流通使用环节无码或者一物多码现象普遍,严重影响了医疗器械生产、流通、使用等各环节对医疗器械的精准识别,难以实现有效监督和管理。
医疗器械唯一标识(Unique Device Identification,简称UDI)是医疗器械的身份证,医疗器械唯一标识系统由医疗器械唯一标识、数据载体和数据库组成。为每个医疗器械赋予身份证,实现生产、经营、使用各环节的透明化、可视化,提升产品的可追溯性,是医疗器械监管手段创新和监管效能提升的重要抓手,对严守医疗器械安全底线、助力医疗器械产业高质量发展都将起到积极作用。因此,我国医疗器械唯一标识系统建设工作急需开展。
医疗器械唯一标识是国际医疗器械监管领域关注的焦点和热点,2013年,国际医疗器械监管机构论坛(IMDRF)发布医疗器械唯一标识系统指南。同年,美国发布医疗器械唯一标识系统法规,要求利用7年时间全面实施医疗器械唯一标识。2017年,欧盟立法要求实施医疗器械唯一标识,日本、澳大利亚、阿根廷等国家也相继开展相关工作,全球医疗器械唯一标识工作不断推进。
2012年,国务院印发《“十二五”国家药品安全规划》,要求“启动高风险医疗器械国家统一编码工作”。2016年,国务院印发《“十三五”国家药品安全规划》,要求“构建医疗器械编码体系,制定医疗器械编码规则”。2019年,国务院办公厅印发《深化医药卫生体制改革2019年重点工作任务》,要求“制定医疗器械唯一标识系统规则”,经中央全面深化改革委员会第八次会议审议通过,由国务院办公厅印发的《治理高值医用耗材改革方案》中,明确提出“制定医疗器械唯一标识系统规则”。2019年7月,国家药监局会同国家卫生健康委联合印发《医疗器械唯一标识系统试点工作方案》,拉开我国医疗器械唯一标识系统建设序幕。








 苏公网安备32050802011615号
苏公网安备32050802011615号