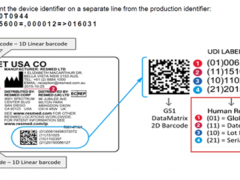
GS是德语"Geprufte Sicherheit"(安全性已认证),也有"Germany Safety" (德国安全)的意思。它于1977年由德国劳工部颁布,以德国产品安全法(GSG)为依据,按照欧盟统一标准EN或德国工业标准DIN进行检测的一种自愿性认证,是欧洲市场公认的德国安全认证标志。
申请GS标志及证书的产品必须由第三方独立认证机构按照欧洲(EN)标准或德国(DIN)标 准执行测试, 同时对生产企业的质量保证体系进行检查,通过后才可颁发证书。 获得GS标志和证书后,申请者可以在产品上打GS标志。认证机构则每年对生产企业执行工厂检查,以保证产品的符合性和一致性。
GS 标志为一项自愿性的测试标志。发给通过测试的产品,标明其它机构已经测试此产品的安全性,并继续维持生产管制。GS 标志表示已经符合德国的工业、进口商、经销商、贸易公司、政府保险及消费者机构的规定,此标志证明科技产品符合「德国产品安全法」的规定。这些机构可能无法自行管理来自全世界产品的一致性,但希望免除产品责任的不确定性及达成客户满意程度。因此 GS 标志的概念相当成功,不仅成为一项营销标准,也广受欧洲及其它国家许多客户及经销商广大的支持。
GS标志表示该产品的使用安全性已经通过公信力的独立机构的测试。GS标志,虽然不是法律强制要求,但是它确实能在产品发生故障而造成意外事故时,使制造商受到严格的德国(欧洲)产品安全法的约束。所以GS标志是强有力的市场工具,能增强顾客的信心及购买欲望。虽然GS是德国标准,但欧洲绝大多数国家都认同。而且满足GS认证的同时,产品也会满足欧共体的CE标志的要求。和CE不一样,GS标志并无法律强制要求,但由于安全意识已深入普通消费者,一个有GS标志的电器在市场可能会较一般产品有更大的竞争力。
GS标志

GS认证产品范围
● 家用电器,比如电冰箱,洗衣机,厨房用具等。
● 家用机械。
● 体育运动用品。
● 家用电子设备,比如视听设备。
● 电气及电子办公设备,比如复印机、传真机、碎纸机、计算机、打印机等。
● 工业机械、实验测量设备。
● 其它与安全有关的产品如自行车、头盔、爬梯、家具等。
GS认证申请形式
(1)首次会议:通过首次会议,检测机构将或代理机构向申请者的产品工程师解释认证的具体程序以及有关标准,并提供将递交要求的文件表格。
(2)申请:由申请者提交符合要求的文件,对于电器产品,需要提交产品的总装图,电气原理图,材料清单,产品用途或使用安装说明书,系列型号之间的差异说明等文件。
(3)技术会议:在检测机构检查过申请者的文件数据后,将会安排与申请者的技术人员进行技术会议。
(4)样品测试:测试将依照所适用的标准进行,可以在制造商的实验室或检验机构的任何一个驻在各国的实验室进行。
(5)工厂检查:GS认证要求对生产的场所进行与安全相关的程序检查。
(6)签发GS证书。
申请GS认证需要的时间和费用
一般来说,时间长短取决于产品是否需要作修改或者生产商提交所需产品文件数据的速度。总的来讲,所需的时间一般约在6~8周。 费用包括一次性发证费,工厂检查费以及证书年费。具体的数目将根据产品的类别以及所需的测试决定。认证机构在收到申请者提交所需要的文件后将为您提供价格参考,每个认证机构的市场政策、公信力不同,价格会有所差异 。
GS与CE的区别
GS:
自愿认证non-compulsory
适用德国安全法规进行检测GS
由经德国政府授权之独立之第三方进行检测并核发GS标志证书
必须缴年费 无须年费
每年必须进行工厂检查
由授权测试单位来核发GS标志, 公信力及市场接受度高
CE:
强制性认证Compulsory
适用欧洲标准(EN)进行检测
在具备完整技术文件(包含测试报告)的前提下可自行宣告CE无须工厂检查
工厂对产品符合性的自我宣告, 公信力及市场接受程度低
GS与LVD的区别
欧共体CE规定, 1997.1.1.起管制 "低电压指令(LVD)"。GS已经包含了“低电压指令(LVD)"的全部要求。所以, 获得GS标志后, TUV会例外免费颁发该产品LVD的CE证明 (COC),TUV Rheinland 97年后的证书则在GS证书中包含了LVD证书。厂商申请GS的同时获得了LVD证明。
申请GS标志及证书的产品必须由第三方独立认证机构按照欧洲(EN)标准或德国(DIN)标 准执行测试, 同时对生产企业的质量保证体系进行检查,通过后才可颁发证书。 获得GS标志和证书后,申请者可以在产品上打GS标志。认证机构则每年对生产企业执行工厂检查,以保证产品的符合性和一致性。
GS 标志为一项自愿性的测试标志。发给通过测试的产品,标明其它机构已经测试此产品的安全性,并继续维持生产管制。GS 标志表示已经符合德国的工业、进口商、经销商、贸易公司、政府保险及消费者机构的规定,此标志证明科技产品符合「德国产品安全法」的规定。这些机构可能无法自行管理来自全世界产品的一致性,但希望免除产品责任的不确定性及达成客户满意程度。因此 GS 标志的概念相当成功,不仅成为一项营销标准,也广受欧洲及其它国家许多客户及经销商广大的支持。
GS标志表示该产品的使用安全性已经通过公信力的独立机构的测试。GS标志,虽然不是法律强制要求,但是它确实能在产品发生故障而造成意外事故时,使制造商受到严格的德国(欧洲)产品安全法的约束。所以GS标志是强有力的市场工具,能增强顾客的信心及购买欲望。虽然GS是德国标准,但欧洲绝大多数国家都认同。而且满足GS认证的同时,产品也会满足欧共体的CE标志的要求。和CE不一样,GS标志并无法律强制要求,但由于安全意识已深入普通消费者,一个有GS标志的电器在市场可能会较一般产品有更大的竞争力。
GS标志
GS认证产品范围
● 家用电器,比如电冰箱,洗衣机,厨房用具等。
● 家用机械。
● 体育运动用品。
● 家用电子设备,比如视听设备。
● 电气及电子办公设备,比如复印机、传真机、碎纸机、计算机、打印机等。
● 工业机械、实验测量设备。
● 其它与安全有关的产品如自行车、头盔、爬梯、家具等。
GS认证申请形式
(1)首次会议:通过首次会议,检测机构将或代理机构向申请者的产品工程师解释认证的具体程序以及有关标准,并提供将递交要求的文件表格。
(2)申请:由申请者提交符合要求的文件,对于电器产品,需要提交产品的总装图,电气原理图,材料清单,产品用途或使用安装说明书,系列型号之间的差异说明等文件。
(3)技术会议:在检测机构检查过申请者的文件数据后,将会安排与申请者的技术人员进行技术会议。
(4)样品测试:测试将依照所适用的标准进行,可以在制造商的实验室或检验机构的任何一个驻在各国的实验室进行。
(5)工厂检查:GS认证要求对生产的场所进行与安全相关的程序检查。
(6)签发GS证书。
申请GS认证需要的时间和费用
一般来说,时间长短取决于产品是否需要作修改或者生产商提交所需产品文件数据的速度。总的来讲,所需的时间一般约在6~8周。 费用包括一次性发证费,工厂检查费以及证书年费。具体的数目将根据产品的类别以及所需的测试决定。认证机构在收到申请者提交所需要的文件后将为您提供价格参考,每个认证机构的市场政策、公信力不同,价格会有所差异 。
GS与CE的区别
GS:
自愿认证non-compulsory
适用德国安全法规进行检测GS
由经德国政府授权之独立之第三方进行检测并核发GS标志证书
必须缴年费 无须年费
每年必须进行工厂检查
由授权测试单位来核发GS标志, 公信力及市场接受度高
CE:
强制性认证Compulsory
适用欧洲标准(EN)进行检测
在具备完整技术文件(包含测试报告)的前提下可自行宣告CE无须工厂检查
工厂对产品符合性的自我宣告, 公信力及市场接受程度低
GS与LVD的区别
欧共体CE规定, 1997.1.1.起管制 "低电压指令(LVD)"。GS已经包含了“低电压指令(LVD)"的全部要求。所以, 获得GS标志后, TUV会例外免费颁发该产品LVD的CE证明 (COC),TUV Rheinland 97年后的证书则在GS证书中包含了LVD证书。厂商申请GS的同时获得了LVD证明。