定义
1. 电磁兼容性
是指车辆、车用部件或独立技术单元在电磁环境下能否满意的工作而且对该环境内的其他部件也不产生不可忍受的电磁干扰的能力。
2. 电磁干扰
是指一种电磁现象,它可以导致车辆、部件或独立技术单元的性能降低。电磁干扰可以是电磁噪声,无用信号和介质传播的变化等。
3. 电磁抗扰度
是指车辆、车用部件或独立技术单元在特定的电磁干扰下保持其性能、继续工作的能力。37Q安规与电磁兼容网
4. 电磁环境是指在给定的场所下存在的总的电磁现象。
5. 参考限值
是指型式批准或判定产品是否符合要求时所使用的指定的电平值。37Q安规与电磁兼容网
6. 基准天线
频率从20至80MHz基准天线是缩短的、半波谐振频率在80MHz的平衡偶极子天线;频率高于80MH z时,是指调谐到测量频率的半波长平衡振子天线。
7. 宽带辐射
是指是指起辐射频带带宽超过了特定的测量仪器或接收机的带宽。37Q安规与电磁兼容网
8. 窄带辐射
是指是指起辐射频带带宽低于特定的测量仪器或接收机的带宽。37Q安规与电磁兼容网
9. 电气/电子系统
是指一个或一组电气电子部件包括其电气连接装置,形成了车辆的一个部分,并且预定与车辆整体进行型式批准。
10. 电气/电子组件(ESA)
是指一个或一组电气/电子部件包括其电气连接装置,它购成车辆的一个部分,执行一个或多个特定的功能。一个ESA可以按照制造商的要求按部件或独立的技术单元(STU)进行批准。(见协议E/ECE/324 第二条:定义)
11 . 汽车类型
就电磁兼容性而言,是指在设计上不能有以下方面本质的差别:
a) 发动机单元的总尺寸和形状;
b) 电气/电子部件的布置及其总线布置;
c) 构成汽车本体或外壳(如果适用的话)的材料,如钢材,铝材或纤维玻璃壳体等;对于汽车面板不同的外观设计,只要本体材料没有发生变化,不影响型式批准,但是这样的改变要通告。
11. ESA类型
就电磁兼容性而言,是指在设计上不能有以下方面本质的差别:
a) ESA所执行的功能;
b) 电气/电子元件的布局(如果适用的话)。
12. ESA型式批准
按照协议E/ECE/324 第三条二款,ESA型式批准申请可由汽车制造商提出,也可由ESA制造商提出。
申请者应按协议E/ECE/324 附录IIB的规定提供充分的信息和文件。
制造商在提交申请时可以提交一份检测报告,型式批准机构可以据此编制批准证书。
13. 型式批准
型式批准程序
车辆的型式批准可以根据制造商的要求选取以下几种方式:
1) 批准车辆的安装
可以按E/ECE/324的第六章直接对车辆的安装进行型式批准。如果车辆制造商选择了此种程序,不需要单独对电气/电子系统或ESA进行检测。
2) 对ESA进行独立检测后对车辆进行型式批准
在此程序下,车辆制造商可以向批准机构出示相关证据,证明:所有的电气/电子系统或ESA均按本协议获得型式批准,并且按证书附件中规定的条件进行了安装,即可获得车辆的型式批准。
3) 如果车辆中没有安装任何可能产生干扰或被干扰的设备,也没有任何电气/电子系统,也没有任何火花点火设备,车辆制造商仍然可以获得(如果他愿意)型式批准,此种型式批准不需要检测。
14. ESA的型式批准
根据ESA制造商的要求,ESA既可以针对所要安装的任何一个车型进行型式批准,也可以针对特定车型或制造商要求的车型。涉及直接控制车辆行驶的ESA通常要按照车辆制造商的协议获得型式批准。
授予型式批准
15. 车辆型式批准
如果代表性的车辆完全满足协议E/ECE/324第六部分的技术要求,则可授予型式批准。
16. ESA
如果代表性的ESA系统完全满足协议E/ECE/324第六部分的技术要求,则可授予型式批准。
17. 为编制车辆型式批准通告书和ESA型式批准通告书,E-mark标志各协议签署国的授权机构可以采用认可的实验室出具的检测报告。
是否可按E/ECE/324授予、不授予车辆、ESA的型式批准都应按照该协议的规定通告该协议的各签约国。
印度尼西亚医疗器械注册要点
印度尼西亚作为东南亚最大的经济体之一,其医疗器械市场近年来展现出强劲的增长势头。印尼的医疗器械市场规模在过去几年中持续扩大,年增长率保持在较高水平。这一增长主要得益于印尼经济的稳定发展、人口的持续增长
0评论2025-05-26276
阿联酋医疗器械注册认证指南
一、基本概况1.自然环境阿拉伯联合酋长国简称阿联酋,位于阿拉伯半岛东部,北濒波斯湾,海岸线长734公里。西和南与沙特阿拉伯交界,东和东北与阿曼毗连。属热带沙漠气候,全年分两季,5至10月为热季,最高气温可达50
0评论2025-05-26214
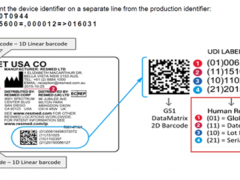
- 欧盟MDR医疗器械注册认证指南
0评论2024-11-14
医疗器械软件注册申报常见问题
问:软件如何进行命名?答:建议在《医疗器械分类目录》《医疗器械通用名称命名规则》和《医用软件通用名称命名指导原则》的框架下,再综合考虑同类已上市产品的名称规范申报注册产品的通用名。问:软件的交付方式有
0评论2024-09-04296
欧盟医疗器械CE注册流程
一、欧盟医疗器械CE注册流程1.你首先需要的是得到一个合格的PRRC(负责法规遵从性的人)。通常由同一个人担任ISO 13485定义的管理者代表。但是,这可以分包给合格的顾问(如CMS)。2.接下来,您需要根据医疗器械法规
0评论2024-09-04200
欧盟发布IVDR延长过渡期的常见问答
2024年7月9日,IVDR过渡期修正案Regulation (EU)2024/1860正式发布。其中更新后第110条讲述了遗留器械的过渡期时限和可以享受过渡期的条件。针对这份延期修正案,欧盟委员会发布了一份《体外诊断医疗器械法规》(IVD
0评论2024-08-09253
欧盟电暖器CE-ErP能效法规更新
法规信息法规名称:(EU) 2024/1103 生效日期:1 July 2025 对象:出口欧盟的电暖器产品主要变化:法规更新取代原法规(EU) 2015/1188更新内容1.管控范围商用局部空间加热器的管控范围由原来的120 kW以下,扩大到300 k
0评论2024-06-18229
浅析欧盟通用充电器的修订指令及指令指导文件
近些年由于ICT 技术的创新和快速发展,市场上的电子设备出现了各种类型的充电接口和充电解决方案,然而由于充电接口规格的不统一、不兼容,一方面带来了大量的电子垃圾,造成了电子资源的浪费,另一方面也给消费者带
0评论2024-06-18146
广东药监局医疗器械常见问题答疑汇总
1、《企业落实医疗器械质量安全主体责任监督管理规定》包含哪些内容?答:《规定》共六章三十条,主要包括三方面内容:一是明确质量安全关键岗位要求。生产企业质量安全关键岗位人员包括企业法定代表人和主要负责人
0评论2024-03-05188
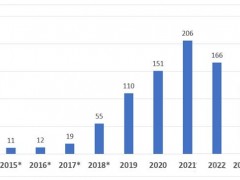
- FDA突破性医疗器械项目常见问题汇总
0评论2023-12-18