文件的起草、准备和保存是欧盟新方法指令要求要加贴CE标志的产品的制造商必须开展的一项工作。新方法指令规定,符合基本要求的产品,在加贴CE标志前,制造商就有责任准备一套完整、充分、符合要求的文件,用以证明或声明投放市场或投入使用的产品符合新方法指令规定的相关基本要求,同时它也是欧盟成员国国家主管当局监督查验的重要内容。文件的质量同产品的质量一样受到监督管理当局的重视。因此,制造商在产品出口前必须按照指令的要求准备一套高质量的、完整的文件,以免给产品出口带来不必要的麻烦和损失。
1 文件的范围和内容
新方法指令要求的文件提供的信息主要用以证明或声明产品符合新方法指令规定的相关基本要求。文件主要包括必要的技术文件、EC合格声明、质量保证文件及其它相关证明文件等,这些文件应能充分反映产品满足了基本要求的信息,反映为使
产品满足基本要求而采取的包括技术和管理措施,以及制造商具有各种保证能力等。其中技术文件是非常关键的,它主要涉及产品的设计、制造和操作等。新方法指令针对不同的产品和适应不同的合格评定程序要求规定了不同的文件要求,包括要求从产品的设计、生产等各个方面提供技术证据。下面就产品在进行CE标志认证中可能会涉及的文件进行的描述,以便在具体的文件准备时参考之用。这些文件中有些是指令要求必须具备的,是国家主管当局开展监督活动的依据;有些虽然不是指令要求的,但它可以有助于证明产品满足基本要求,国家主管当局在市场监督进行风险评估时会以非歧视的方式予以考虑,如产品认证、质量体系的建立等。
1.1 设计文件
产品设计是决定产品质量的关键环节,产品能否满足新方法指令规定的基本要求,关键在于产品设计时是否遵循了和满足了这些要求,而这些信息就是通过一系列的技术文件予以表达的。这些技术文件一般包括:
(1)规定了产品设计应遵守的基本要求、标准和其它技术规范、或为满足基本要求而规定了要采取的措施等的设计输入文件。如产品设计或技术任务书、产品采用标准要求等;(2)用以体现产品满足了基本要求或贯彻了相关技术措施及有助于产品安装、使用、维护等的设计图样。如产品的结构图、安装图、电器系统图、液压系统图及其它动力源有关部门的图纸等;(3)作为设计依据或验证的技术文件,如设计计算书、验算书、技术分析文件等;(4)用以指导用户正确安装、适宜维护保养、正确操作以及明示或警示存在危险或可能存在的危险的技术文件,如产品安装说明书、产品操作使用及维护说明书、安全使用手册等。
1.2 危险分析及采取的措施文件
新方法指令所规定的是保护公众利益的基本要求,而基本要求是针对特定产品可能潜在的危害性,因此,制造商必须能够通过分析,识别产品可能潜在的全部危害性及这些危险的严重程度,以便确定产品必须满足的基本要求和应采取的措施,来消除可能的一切事故风险。同时,制造商应将这些分析、评价和采取的措施文件化,以证明产品能够满足新方法指令规定的相关基本要求。这些文件包括对产品的可能存在的潜在危险进行分析和评价的文件,以及为消除危险所采取的措施的文件和用以证明这些措施有效性的确认文件等。
1.3 生产文件
对于连续或成批生产的产品,为保证后续生产的产品能够和通过CE标志认证的产品的一致性,制造商应在产品的生产制造方面采取必要的技术和管理措施,保证产品的生产过程不会对产品满足新方法指令的基本要求造成不良影响,并将这些措施文件化,以便能够持续贯彻。同时,通过这些文件化的措施证明制造商有能力确保所提供的产品始终符合设计的要求(与样机一致)。这些文件包括制造商采取的工艺措施、质量管理措施等,如对关键零部件的加工采取的特殊工艺措施文件、对外购原材料和零部件的质量控制措施文件、生产过程的管理和控制文件、产品检验管理控制文件等。
1.4 符合性验证和证明文件
符合性验证和证明文件主要是指那些能够直接或间接证明产品或零部件满足新方法指令所规定的相关基本要求的文件。
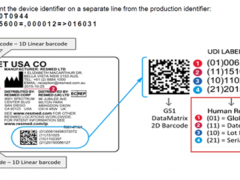
(1)产品中使用的新方法指令覆盖的产品(包括设备、仪器、器具、用具、材料、组件、部件或安全部件、元件、连接件、附件或系统),除了产品上应有CE标志外,还应取得这些产品符合新方法指令基本要求的证明材料,如该产品的EC合格声明、EC型式检验合格证书或必要技术文件等文件复印件;
(2)产品中可能会对整机安全或环保性能产生影响的主要零部件(不属于新方法指令覆盖范围的),尽量索取由权威机构出具的检验或试验报告,或者是该零部件已获得的安全认证证书及其它可能的相关证书等的复印件;
(3)制造商对零件、配件或机器整体进行的必要研究或试验,以确定机器在这种设计或制造方式下能否安全的安装起来并交付使用的文件,如试验报告、研究报告等;
(4)即使指令没有要求,制造认为需要,也可提供从具有法定资格的权威机构及实验室获得的该产品的技术报告或证书;
(5)对于新方法指令要求有第三方(欧盟指定的公告机构)参与进行合格评定的,应有公告机构出具EC型式检验报告和EC型式检验合格证书;
(6)制造商起草、签署的EC合格声明。
1.5其他文件
除了上面所列的四个方面的文件外,一般还应形成和保持下列文件:
(1)企业基本情况信息的文件;(2)一份包含产品符合的新方法指令的基本要求、采用的标准及采用的其它技术规范的文件清单;(3)产品应满足的新方法指令的基本要求、采用的协调标准及采用的其它标准和技术规范等文件;(4)如果制造商任命了自己的在欧共体授权代理,应建立的明确界定任务的内容和代理权限的书面授权书;(5)如果产品适用的是以ENISO9001、9002或9003标准为基础的合格评定模式,制造商为满足新方法指令所规定的要求,在实施和应用质量体系时,应把对产品符合指令所规定的基本要求纳入质量目标、质量策划、质量手册和控制文件中,并使采用的协调标准或确保产品满足基本要求的技术方法措施文件化。