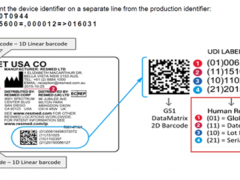
E/e Mark 也就是欧洲共同市场 , 对汽.机车及其安全零配件产品,噪音及 废气等,均需依照欧盟法令【 EEC Directives 】与欧洲经济委员会法规【 ECE Regulation 】的规定,通过产品符合认证要求,即授予合格证书,以确保行车的安全及环境保护之要求。
E-Mark 依 认证国 别不同,所授予之编号也不同,例如:向卢森堡提出申请,其 E-Mark 标志为 E13/e13。
适用产品之范围
整车-即两轮或三轮以上之电机动交通工具,如客车、货车、摩托车、巴士及道路外之车辆
汽机车零组件-车灯与灯泡、各种视镜、轮胎、轮圈、剎车、喇叭、防盗设备、安全带、汽车玻璃及排气管等
汽机车零配件-安全帽、儿童安全椅、车内附属电器产品等
自 2002年10月起,规定所有车辆,车辆零部件,以及用于车上的电子性产品必须
强制执行EMC测试。 所有在欧销售的电子零部件须统一符合EMC指令95/54/EC,根据EMC指令89/336/EEC进行的自我宣告将不再有效。 而由欧盟授权车辆类产品的公告机构出具E/e Mark证书。也就是说,车辆类电子及电子零部件原先申请的CE(EMC)认证将从2002年10月起不再有效。 必须重新申请欧洲国家交通部门出具的E/e Mark证书后方可在欧洲市场销售。
欧洲对于机动车整车及涉及安全的零部件和系统有安全认证的要求﹐具体体现为 E标志和e标志认证.
E标志源于欧洲经济委员会(Economic Commisssion of Europe, 简称ECE)颁布的法规(Regulation)﹒目前ECE包括欧洲28个国家﹐除欧盟成员国外﹐还包括东欧﹐南欧等非欧国家﹒ECE法规是推荐各成员适用﹐不是强制性标准﹒成员国可以套用ECE法规﹐也可以延用本国法规﹒目前从市场需求来看﹐通常ECE成员愿意接收符合ECE法规的测试报告及证书﹒E标志证书涉及的产品是零部件及系统部件﹐没有整车认证的相应法规﹒获得E标志认证的产品﹐是为市场所接受的﹒国内常见E标志认证产品有汽车灯泡﹐安全玻璃﹐轮胎﹐三角警示牌﹐车用电子产品等﹒E标志认证的执行测试机构一般是ECE成员国的技术服务机构﹒E标志证书的发证机构是ECE成员国的政府部门﹒各国的证书有相应的编号﹕
E1—德国 E2—法国 E3—意大利 E4—荷兰 E5—瑞典
E6—比利时 E7—匈牙利 E8—捷克 E9—西班牙 E10—南斯拉夫
E11—英国 E12—奥地利 E13—卢森堡 E14—瑞士 E16—挪威
E17—芬兰 E18—丹麦 E19—罗马尼亚 E20—波兰 E21—葡萄牙
E22—俄罗斯 E23—希腊 E25—克罗地亚 E26—斯洛文尼亚 E27—斯洛伐克
E28—白俄罗斯 E29—爱沙尼亚 E31—波黑 E37—土耳其
e标志是欧盟委员会依据欧盟指令强制成员国使用的机动车整车﹐安全零部件及系统的认证标志﹒测试机构必须是欧盟成员国内的技术服务机构﹐发证机构是欧盟成员国政府交通部门﹒获得e标志认证的产品各欧盟成员国都将认可﹒同E标志认证一样﹐各成员国的证书有相应的编号﹕
e1—德国 e2—法国 e3—意大利 e4—荷兰 e5—瑞典
e6—比利时 e9—西班牙 e11—英国 e12—奥地利 e13—卢森堡
e17—芬兰 e18—丹麦 e21—葡萄牙 e23—希腊 E24—爱尔兰
无论是 E标志或e标志认证﹐首先产品要通过测试﹐生产企业的质量保证体系也要至少达到ISO9000标准的要求 。 本公司可为生产企业提供认证前的标准辅导﹐样品部分预测﹐大大提高了通过率 。 我们高效﹐快捷﹐专业的服务﹐不仅仅是为您取得认证﹐还包括以后的厂检辅导﹐标准更新﹐证书更新﹐出货检查等方面﹐以保证贵公司产品在欧洲受到普遍的认可 。
关于车辆和车辆电气/电子部件按协议E/ECE/324进行E-Mark 型式批准的有关电磁兼容性定义和标志要求
注:E-Mark 型式批准依据协议E/ECE/324,e-Mark型式批准依据指令95/54/EEC,两种型式批准在定义和技术要求方面基本相同,不同的是相互认可的国家范围不同,标志及标志的使用管理不同。前者相互认可的国家更多。以下介绍是针对我检测中心授权开展的E—Mark检测所涉及的内容。