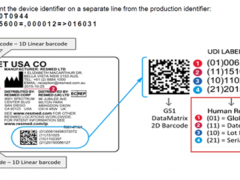
电信设备专家评审组由科研、检测、运营和政府等部门的专家组成,并根据不同的专业领域,分为传输组、接入组、交换/信令/智能网组、移动组、数据组五个不同的专家小组。专家组成员应符合如下条件:①坚持原则,客观公正,具有良好的科学道德和职业道德;②精通专业业务,熟悉产品/技术/标准情况,在其专业领域享有一定声誉;③熟悉电信设备进网相关政策,并能积极参与相关活动;④不在生产企业任职或兼职,不向生产企业提供与评审设备有关的技术咨询服务。参加评审的专家对被评审设备的技术负有保密义务,不得窃取或泄漏生产企业的技术秘密。
一、需要进行专家评审的电信设备分类
在评审目录中的电信设备,按照其采用的技术、标准的成熟程度以及对通信网的影响,需要进行专家评审的电信设备可以分为如下四类:
●A类——设备技术、标准成熟,一般可以由申请单位对该设备的技术指标进行描述,并提出免评审的申请。
●B类——设备技术、标准成熟,超过一定的应用规模时需评审。
●C类——设备已有标准,但技术仍在发展,在采用新技术或提供新型业务时需评审。
●D类——设备技术、标准不十分成熟,需评审。
评审目录和分类情况见附录一。
二、专家评审的资料要求
①进网许可申请表。
②生产企业介绍。
③总体技术方案,应包括产品概述、设计依据与执行标准、系统组成和功能框图、系统的软硬件结构、系统支持的业务、接口及兼容性、设备的性能和技术指标、系统的组网能力、可靠性设计及环境适应性、操作维护管理。
④进网检验报告,包括电磁兼容测试报告(仅对需要进行电磁兼容测试的设备)。
⑤进网试用、试验报告;申请进网的设备应在地市级运营商电信网上试运行三个月以上或在模拟试验网上试运行一个月以上。试用、试验报告由试验单位出具,报告中应包括设备名称、型号,试验的时间、地点;设备安装的容量、实际用户数;组网结构图;该设备提供的业务;一致性及兼容性测试内容和数据;故障情况及解决措施;用户反映情况;试验单位意见等内容。
⑥质量保证体系认证证书。
⑦售后服务措施。
⑧无线电发射设备型号核准证(仅对发射无线电信号的无线电通信设备)。
三、专家评审的主要内容
①专家审核进网设备的技术方案和实现方法,判断是否具有先进性?
②审核进网设备的质量检验报告和试用报告,对报告中反映出来的问题进行询问,判断其是否影响设备的正常运行以及对相关的设备是否有不良影响?
③进网设备是否存在不允许有的功能和协议等?
④进网设备是否具备应有的功能和协议等?
⑤是否存在应该支持但是没有支持或没有检验合格的接口、功能等,对进网使用是否有影响?
⑥是否有标准中没有列出的功能和接口等?
⑦进网设备的安全性能如何?
⑧对运营商开展的业务支持能力如何?
⑨生产厂家的售后服务体系如何,质量保证措施是否齐备?
四、专家评审程序
每月5日前确定当月的专家评审计划,申请单位应提前三天将评审资料报至电信设备认证中心进行初步审核。评审会原则上安排在每月的10~20日,遇节假日顺延。具体的步骤如下:
①在申请单位提出电信设备进网许可申请后,电信管理局将书面告知申请单位该设备是否需要评审。
②需要评审的电信设备在完成进网检验、质量保证体系审核和试运行后,电信设备进网受理机构报电信管理局,提请进行专家评审。
③电信管理局对所报材料确认后,由电信设备认证中心通知申请单位准备参加专家评审会,并按照要求准备资料。
④电信设备认证中心通知专家并组织评审会。
⑤评审会后,专家组的意见随其他申请资料送交电信管理局进入后续审批流程。