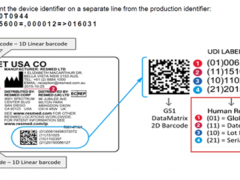
LED驱动电源,2014年以前属于CQC自愿性认证范畴。现在纳入CCC认证名录,照明电器类别镇流器种类项下增加LED模块用直流或交流电子控制装置产品,其中的意义十分明显。
执行标准《强制性产品认证实施规则照明电器》第12项LED模块用直流或交流电子控制装置作为LED驱动电源(以下都以此作为LED模块用直流或交流电子控制装置的简称)的认证依据,它执行的国标分别是:
A,安全标准:
1)GB 19510.1 -2009 /IEC61347-1:2007
灯的控制装置第1部分:一般要求和安全要求;
2)GB 19510.14 -2009 / IEC61347-2-13:2006
灯的控制装置第14部分:LED模块用直流或交流电子控制装置的特殊要求。
B,电磁兼容标准:
1)GB17743-2007 / CISPR 15 :2005
电气照明和类似设备的无线电骚扰特性的限值和测量方法;
2)GB17625.1 -2003 / IEC61000-3-2:2001
电磁兼容限值谐波电流发射限值(设备每相输入电流16A)。
ODM(Original Design Manufacturer)生产厂:利用同一质量保证能力要求,生产过程控制及检验要求及同一产品设计生产相同产品的组织。
ODM初始认证证书持证人:初次获得ODM产品认证证书的组织。
OEM(Original Equipment Manufacturer)生产厂:按委托人提供的设计、生产过程控制及检验要求生产认证产品的生产厂。委托人可以是认证委托人或生产者(制造商);OEM生产厂根据委托人提供的设计、生产过程控制及检验要求,在OEM生产厂的设备下生产认证产品。
一、初始CCC认证证书的ODM模式
首先,生产者(制造商)和生产企业(生产厂)为同一企业,通过自有产品设计、生产过程控制及检验要求等生产相关产品,且生产的产品未获得过强制性产品认证;其次,CCC申请人直接使用上述未获证产品进行CCC认证申请的模式。
二、利用已获证结果获取CCC证书的ODM 模式
利用现有CCC证书(证书状态为正常、生产企业(生产厂)不能为D类企业)作为基础证书,以仅变更基础证书CCC认证委托人(或型号命名)的方式获取新证书的3C认证申请模式(新证书不得再作为基础证书使用)。
注:以ODM/OEM 委托CCC认证的,未提供有效的ODM协议书、OEM协议书、授权书及相关证书复印件。
三、以ODM模式进行CCC认证申请时的送样要求
以初始CCC认证证书的ODM 模式申请时,送样试验的样品由认证委托人按CQC/实验室的要求选送代表性样品用于检测。以利用已获证结果取得3C证书的ODM模式申请时,认证工程师需对CCC认证委托人提供的相关资料(如ODM协议、授权书、型号对照表等)进行核查,如不能确定本次申请型号与原获证型号是否一致,可下达送样通知,由认证委托人将样品送往实验室进行核查。
四、ODM/OEM模式的CCC工厂检查要求
利用已获证结果取得ODM证书的情况(以初始认证证书模式取得的ODM证书的工厂检查要求同新申请)
ODM模式初始工厂检查:利用已获证结果取得ODM证书时无需进行初始工厂检查。
年度监督检查:
对ODM 证书的监督检查随ODM生产厂(生产企业)的监督检查一起进行,检查内容包括ODM合作协议的执行情况、3C认证标志管理、顾客产品管理、生产销售管理、ODM生产厂(生产企业)为其他生产者(制造商)生产认证产品的实际情况等。在进行一致性检查时应特别关注ODM产品的一致性。
OEM模式初始工厂检查:
根据该申请对应的CCC认证模式判定是否需要进行初始工厂检查。主要查采购与关键元器件和材料控制、生产过程控制、例行检验/确认检验和现场指定试验、CCC认证产品的一致性要求等条款及产品一致性检查,但不排除对其它必要和/或质疑条款进行重新检查确认。
年度监督检查:
OEM证书的年度监督检查同新申请。
注:OEM工厂检查时,需额外提供如下资料:OEM合同;相关授权文件(如CCC标志在OEM工厂使用的授权文件等)。
五、ODM变更的特殊要求
ODM认证产品变更申请须初始CCC认证证书持证人提出,经批准后,其他ODM认证证书持证人须在1 个月内提交认证变更申请。但不涉及安全和电磁兼容(如申请人名称、产品型号命名方式、证书有效期等变更)的除外。