


医疗器械关系到生命和健康,既有合规要求也有质量要求。产品设计与开发阶段至关重要,优质产品是通过科学设计实现的。合规和质量的要求需要植入到设计开发过程,在产品孕育之初打好基础。文章对设计开发和注册过程进行研究和分析,提出将集成产品开发和质量管理体系融合,厘清了设计开发评审、验证和确认的三者关系,明确了设计开发进度、成本和质量的主次联系。
关 键 词:医疗器械、研发质量、集成产品开发、质量管理、产品注册
医疗器械需要满足“安全、有效和质量可控”。医疗器械用于保障人的生命和健康安全,中国、美国和欧盟等国都出台了一系列法律法规和标准,监管部门严格把关医疗器械产品的上市门槛,满足合规底线要求。对于医疗器械,既要合规,又要保证产品质量,两者缺一不可[1]。研发作为产品的源头部门,例如原材料清单,验证和确认资料,量产后生产作业指导书等文件都是来源于研发部门,所以医疗器械质量和合规工作要从研发抓起,合规和质量策划是连接合规要求、质量目标和具体的管理活动之间的桥梁纽带。
1.医疗器械设计开发的重要性
医疗器械的开发过程复杂而漫长,需要不同专业背景岗位的协助配合,如电路设计、软件设计,人体工程学、物理治疗、化学分析、临床知识、卫生经济学等;同时除了产品本身设计开发的时间,还包含了注册审评的时间,进一步延缓了产品的上市节奏。该行业是集知识密集型、资金聚集型、多学科交叉、质量要求高等诸多特点于一身的高科技产业,但我国产品产品以中低端为主,生产和研发投入不足[2]。
该行业门槛较高,属于政府强监管行业,对产品的安全、有效提出更高的要求。中国医疗器械监督管理基本原则是风险管理,全程监控,科学监管,社会共治的基本原则。中国医疗器械监管开启了4G年代,有生产规范、临床试验规范、经营规范、使用规范、医疗法规贯穿了产品的整个生命周期[3]。
研发是伴随着产品从生到死的一个核心部门,包含产品的需求导入,产品的硬件和软件的实现,产品的验证,临床的确认,产品的注册,上市后的不良事件,产品的退市等工作。政府部门的监管工作同样是聚焦医疗器械产品的全生命周期管理。
产品研发之初就要考虑到上市区域的法律法规要求,注册路径等。在研发需求输入阶段时候,就需要考虑产品的标准要求,可用性工程要求,软件质量要求,临床确认要求,网络安全的要求,包装要求,无菌要求等。产品研发样机形成后,需要开展设计转化工作。研发部门需要形成产品原材料清单,生产作业指导书,检验操作规程,以及各种工装夹具,相关文件的配套表单等。产品投入市场后,如果涉及不良事件,产品变更,质量问题,都需要研发部门的深度参与。
无数的企业投入大量的人力、物力进行研发,最终却没有得到商业回报,例如研发进度缓慢,定价过高,验证和确认不充分不能通过监管部门的审评,或者体系核查弄虚作假,根本原因在于思想上没有认识到研发管理的重要性,在需求阶段未深入调研市场需求,合规和质量等要求。
2. 注册监管要求
2.1 注册申报资料来源于设计开发阶段
在进行产品注册申报过程(见图1)中,要提交产品技术要求,产品风险分析报告,临床评价报告,检测报告,产品说明书和标签样稿,以及使用寿命验证报告等文件,这些文件都是在研发过程中形成初版,并在过程中升版修订,形成最终版。在产品立项之初,需要将该产品所适用的法规(例如涉及产品分类,质量体系要求,产品注册指导原则,临床试验质量要求等法规),标准(例如电气安全,电磁兼容,可用性,清洁消毒,生物相容性等标准),质量指标(例如关键性能参数,试产直通率,缺陷关闭率等指标)作为输入纳入到产品需求包中。
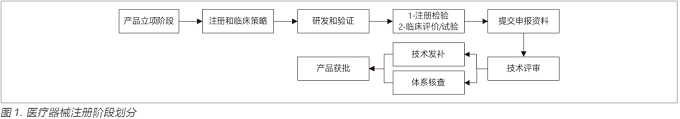
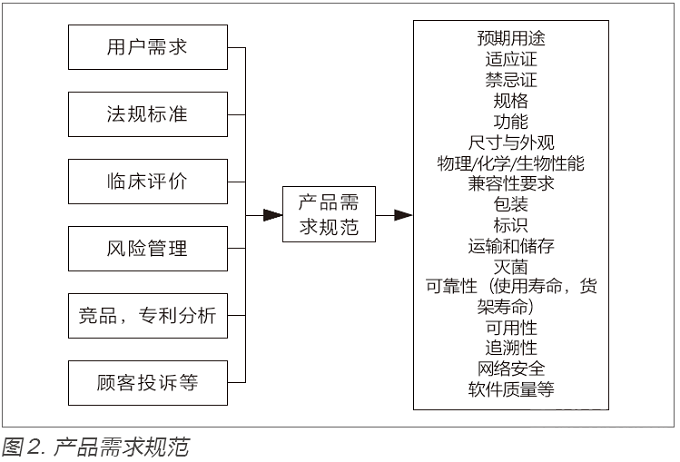
需求包不仅仅包含法规,标准,临床评价,风险管理要求,还包含用户需求,竞品和专利分析等。通过这些不同方面的输入,形成了产品需求规范,转化成产品的各个要素,这些要素要求正是注册文档的源头(见图2)。
当这些需求形成后,进入设计开发环节,在每个环节均要有交付物,含文档和研发样机(或模块)。这些交付物需要质量体系工程师,法规工程师,技术专家进行评审,通过后设计开发才能进入下一环节。结合图1注册阶段的划分,产品全生命周期每个环节需要开展的主要工作,见图3。

2.2 注册核查的重点在于设计开发阶段
国际标准ISO 13485《医疗器械 质量管理体系用于法规的要求》,中国《医疗器械生产质量管理规范》(以下简称《规范》)和《医疗器械注册质量管理体系核查指南》(以下简称《核查指南》)都强调了研发的重要性。
《规范》第六章设计开发的内容,与ISO13485 第七章内容比较类似,介绍了设计开发的过程,含策划、输入、输出、评审、验证、确认、转换,更改的控制和文档管理[4]。
在《核查指南》第4.5 设计开发节中,对设计开发的要求更加具体,特别重视研发过程中形成的记录文件和原始数据资料[5]。例如:①医疗器械设计和开发文档应当源于设计开发策划、输入、输出、评审、验证、确认、转换、变更的相关文件,包含设计开发过程中建立的记录,应当确保历次设计开发最终输出过程及其相关活动可追溯;②申请人应当保留产品设计转换活动的所有记录,以表明设计和开发输出成为最终产品规范前已得到充分验证且适用于常规生产,并确保生产工艺在使用确定的原材料和设备条件下,持续稳定生产出符合预期用途和产品技术要求的产品;③应当保存设计和开发验证活动的详细原始数据记录资料,包括验证方案、验证报告、验证记录(如测试数据、样品处理记录等)、辅助记录等。






 苏公网安备32050802011615号
苏公网安备32050802011615号